
Designing soluble versions of dementia-related proteins
In July 2025, I attended the Alzheimer's Association International Conference as part of the 2025 IBB NextGen event. There, I deepened my understanding of pathogenic proteins such as amyloid beta, tau, and alpha-synuclein. At the time, I had just used the QTY code, a protein design protocol, to create soluble versions of opsins. Inspired by this, I considered using this code to design soluble dementia-related proteins in order to break protein aggregates. However, the idea did not work as planned. In this blog, I will share some of the investigations I conducted.
Tau
I used AlphaFold as my main tool of protein structure prediction. When I attempted to predict the structure of multiple tau proteins, either native or QTY-designed, AlphaFold returned "Model inference failed", and I was unable to proceed. This may be due to the existence of certain highly disordered regions.
Alpha-synuclein
I identified the core aggregate-forming region of alpha synuclein, which consists of 140 amino acids. All the following investigations were carried out on this truncated version of the protein.
First, I used the QTY code to design a soluble version of the protein. The QTY code substitutes hydrophobic amino acids (L, I/V, and F) with similar-shaped, polar, hydrophilic amino acids (Q, T, and Y). This reduces the protein's surface hydrophobicity while preserving the overall structure. In the following image, the upper sequence is native alpha-synuclein and the lower one is the QTY-designed version.

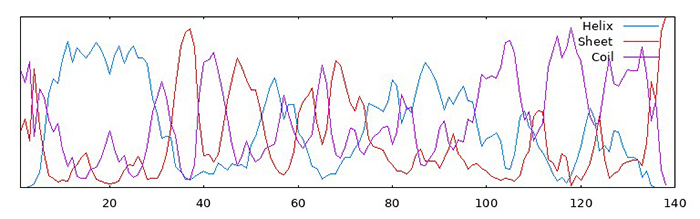
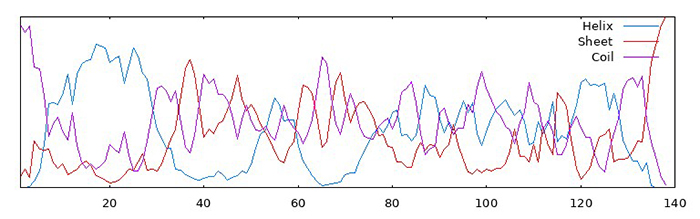
Then, I used GOR to predict the secondary structures of both native and QTY-designed alpha-synuclein. The first image shows the results for the native protein, and the second for the QTY-designed version. It can be seen that the probability of forming β-sheets have decreased. Since the aggregations are in the form of β-strand helices, this might indicate that QTY-designed versions have lower probability of aggregation.
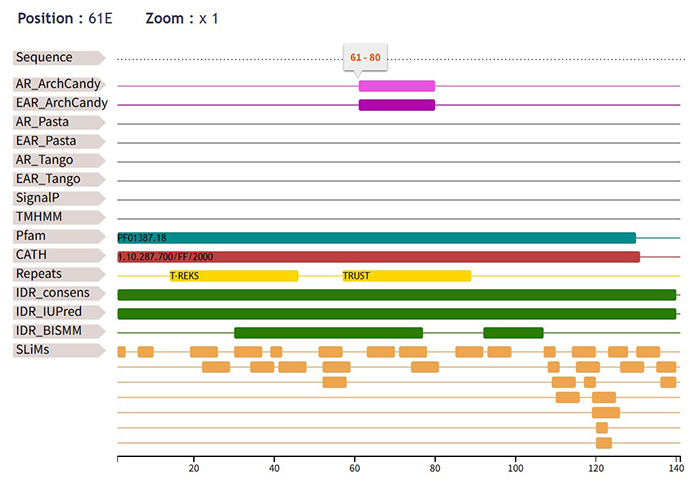
However, TAPASS predicts that there is an aggregation-prone region in the QTY-designed alpha-synuclein, as shown in this figure:
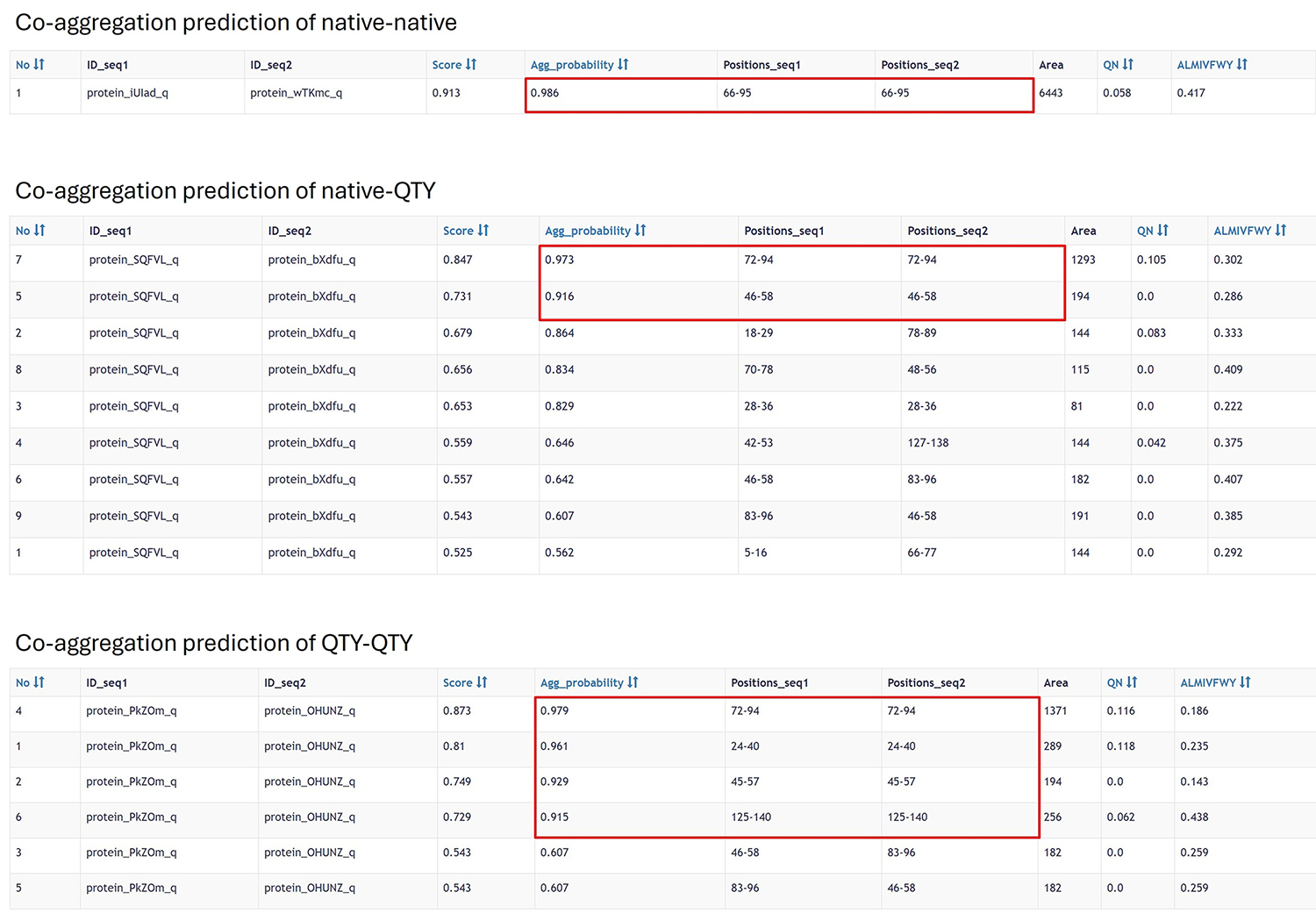
AmyloComp also predicts a high possibility of co-aggregation, both among QTY-designed proteins themselves and between native and QTY-designed proteins:
Finally, AlphaFold also demonstrates the formation of aggregates when multiple copies of native and QTY-designed alpha-synuclein are present:
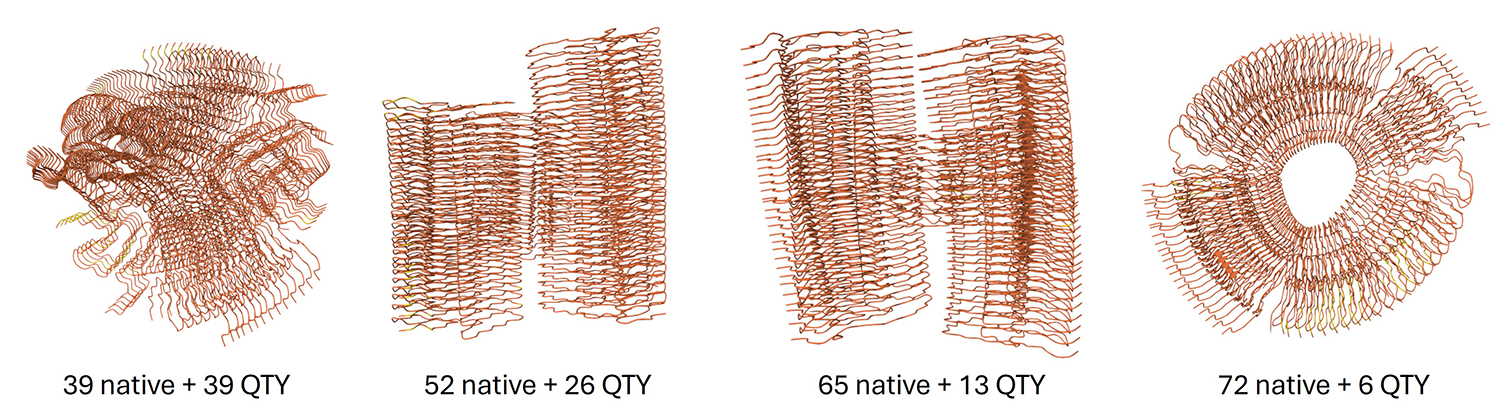
Although previous QTY-related studies have shown that the QTY code works well with β-barrels in bacterial proteins, it is not working so well with the β-helix. I suspect this may be due to structural differences:
- In the β-barrel, the β-strands are aligned inside the cylindrical "surface", so that the residues stick out perpendicularly, directly contacting water.
- However, in the β-helix, the β-strands are perpendicular to the planar "surface" occupied by each protein, so that the residues are inside the plane. Therefore, residue substitutions have limited effects on interactions between adjacent proteins.
If this is the case, then QTY substitutions will not help break alpha-synuclein aggregates.
However, we should also consider that although QTY-designed alpha-synuclein does not modify its interaction with water on the outer surface, it may allow water to enter the aggregate, providing a force to disrupt the aggregate. Indeed, the AlphaFold predictions are all shown in orange, with low certainty. The other programs may also be unreliable, since they are based on existing aggregate-prone proteins, but QTY-designed proteins may be unlike any that is observed in nature. We are in need of computational tools that can more accurately describe the dynamics of aggregate formation.
In conclusion, my investigations suggest the QTY-designed alpha-synuclein is unlikely to be able to break native alpha-synuclein aggregates. However, conducting a wet-lab experiment would be the most efficient and definitive method to confirm the result.
References
These are some of the main sources that I referenced:
- Zhou, Y., Kloczkowski, A., Faraggi, E., & Yang, Y. (Eds.). (2017). Prediction of Protein Secondary Structure (Vol. 1484). Springer New York. https://doi.org/10.1007/978-1-4939-6406-2
- Navarro, S., & Ventura, S. (2022). Computational methods to predict protein aggregation. Current Opinion in Structural Biology, 73, 102343. https://doi.org/10.1016/j.sbi.2022.102343
- Prabakaran, R., Rawat, P., Thangakani, A. M., Kumar, S., & Gromiha, M. M. (2021). Protein aggregation: In silico algorithms and applications. Biophysical Reviews, 13(1), 71–89. https://doi.org/10.1007/s12551-021-00778-w
The following is the citations of the tools I used, including GOR, TAPASS, AmyloComp, and AlphaFold:
- Sen, T. Z., Jernigan, R. L., Garnier, J., & Kloczkowski, A. (2005). GOR V server for protein secondary structure prediction. Bioinformatics, 21(11), 2787–2788. https://doi.org/10.1093/bioinformatics/bti408
- Falgarone, T., Villain, É., Guettaf, A., Leclercq, J., & Kajava, A. V. (2022). TAPASS: Tool for annotation of protein amyloidogenicity in the context of other structural states. Journal of Structural Biology, 214(1), 107840. https://doi.org/10.1016/j.jsb.2022.107840
- Bondarev, S. A., Uspenskaya, M. V., Leclercq, J., Falgarone, T., Zhouravleva, G. A., & Kajava, A. V. (2024). AmyloComp: A Bioinformatic Tool for Prediction of Amyloid Co-aggregation. Journal of Molecular Biology, Computation Resources for Molecular Biology, 436(17), 168437. https://doi.org/10.1016/j.jmb.2024.168437
- Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A. J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C.-C., O’Neill, M., Reiman, D., Tunyasuvunakool, K., Wu, Z., Žemgulytė, A., Arvaniti, E., … Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(8016), 493–500. https://doi.org/10.1038/s41586-024-07487-w